Культивирование вирусов
| Загрузить архив: | |
| Файл: ref-9035.zip (25kb [zip], Скачиваний: 62) скачать |
ОГЛАВЛЕНИЕ
Введение...............................................................................................................................3
1. Виды культур клеток........................................................................................................4
2. Питательные среды..........................................................................................................6
3. Получение клеточных культур...............................................................................…9-18
3.1. Приготовление первичных клеточных культур………………………................…9
3.2. Получение монослойных перевиваемых культур клеток......................................10
3.3. Роллерное культивирование клеток.......................................................................11
3.4. Суспензионное культивирование клеток...............................................................13
3.5. Культивирование клеток на микроносителях……………………………………..16
4. Выращивание вирусов в культурах клеток..............................................................19-24
4.1. Предосторожности при работе с зараженными вирусами клетками....................20
4.2. Обнаружение вирусов.............................................................................................21
4.3. Культивирование пикорнавирусов на примере получения полиовируса
типа 3 в культуре клеток HeLa................................................................................24
Список литературы.............................................................................................................25
ВВЕДЕНИЕ
Для количественного накопления вирусов наиболее удобную систему представляют культуры клеток. Первые попытки культивирования клеток животных вне организма относятся к концу прошлого столетия. Эти отрывочные наблюдения указывали на возможность сохранения жизнеспособности тканей и клеток в искусственных условиях и положили начало глубоким научным исследованиям тканевых культур.
Большая заслуга в деле paзработки методов культивирования тканей принадлежит Каррелю Он впервые доказал возможность размножения клеток животных в искусственных условиях и тем самым продемонстрировал их «бессмертность» и сходство с одноклеточными свободноживущими организмами. Значительных успехов в этом направлении достигла группа исследователей под руководством Эрла. Они первые получили рост большого числа клеток на стекле и в жидкой перемешиваемой суспензии. Появление антибиотиков и успехи в создании искусственных питательных сред открыли новую эру в развитии методов тканевых культур.
Тканевые культуры давно нашли применение для решения различных вопросов биологии и медицины. Однако лишь успехи в области вирусологии, достигнутые с помощью тканевых культур, явились мощным стимулом их развития до современного уровня.
Культивирование вирусов помогает решить ряд теоретически проблем, связанных с изучением особенностей взаимодействия "вирус-клетка". Кроме того решение целого ряда прикладных задач, связанных с диагностикой и производством препаратов для профилактики вирусных инфекций невозможно без накопления вируссодержащего сырья.
1. ВИДЫ КУЛЬТУР КЛЕТОК.
Перенесение клеток целостного организма в условия жизни invitroпрекращает существование их как одного из многочисленных структурных элементов ткани или органа, в состав которых они ранее входили. При этом клетки выходят из-под контроля нейро-гуморальных факторов и приобретают ряд особенностей, зависящих как от самого факта отторжения клеток от этих тканей, так и от конкретных условий их существования in vitro.
Живущие вне организма клетки или ткани характеризуются целым комплексом метаболических, морфологических и генетических черт, резко отличных от свойств клеток органов и тканей in vivo.
В зависимости от методики первичной эксплантации ткани и техники ее культивирования различают несколько видов переживающих и растущих культур тканей и клеток. Наибольшее распространение имеют однослойные, а также суспензионные культуры растущих клеток. Именно они составляют основу современной лабораторной и производственной вирусологической практики.
Различают два основных вида однослойных клеточных культур: первичные и перевиваемые.
Термином «первичная» обозначают клеточную культуру, полученную непосредственно из тканей человека или животных в эмбриональном или постнатальном периоде. Срок жизни таких культур ограничен. По прошествии определенного времени в них возникают явления неспецифической дегенерации, что выражается в грануляции и вакуолизации цитоплазмы, округлении клеток, утрате связи между клетками и твердым субстратом, на котором они выращивались. Периодическая смена среды, изменение состава последней и другие процедуры могут лишь несколько увеличить сроки жизни первичной клеточной культуры, но не могут предотвратить ее конечной деструкции и гибели. По всей вероятности, этот процесс связан с естественным угасанием метаболической активности клеток, выведенных из-под контроля нейро-гуморальных факторов, действующих в целостном организме.
Лишь отдельные клетки или группы клеток популяции на фоне дегенерации большей части клеточного пласта могут сохранить способность к росту и размножению. Эти клетки, обнаружив потенцию бесконечного размножения in vitro, при многократных перевивках дают начало перевиваемым культурам клеток.
Различают линии и штаммы перевиваемых клеток. Первым термином обозначают перевиваемые клетки, характеризующиеся потенциальным бессмертием и, как правило, гетероплоидным кариотипом вторым термином — полуперевиваемые клетки с диплоидным набором хромосом и ограниченной продолжительностью жизни in vitro. Появление и тех, и других клеток связано с процессом отбора в клеточной популяции первичных культур, являющихся, таким образом, источником всех линий и штаммов перевиваемых клеток.
Основное преимущество линий перевиваемых клеток, по сравнению с любой первичной культурой, состоит в потенции неограниченного размножения вне организма и относительной автономности сближающей их с бактериями и одноклеточными простейшими.
Способность перевиваемых клеток к бесконечному размножению in vitroзнаменует собой качественный скачок, в результате которого клетки приобретают способность к автономному существованию, подобно микроорганизмам, выращиваемым на искусственных питательных средах. Совокупность изменений, приводящих к появлению у клеток таких особенностей, называют трансформацией, а клетки перевиваемых тканевых культур — трансформированными.
Усовершенствование техники культивирования клеток значительно расширило возможности получения перевиваемых клеточных линий из самых разнообразных тканей животных и человека. При этом не был обнаружен возрастной предел, выше которого ткани утрачивали бы способность адаптации к неограниченному росту invitro, т.е. к трансформации.
Другим источником перевиваемых клеточных линий являются злокачественные новообразования. В этом случае трансформация клеток происходит invivoв результате развития патологического процесса, этиология которого во многом остается еще невыясненной.
Не все злокачественные новообразования способны давать начало перевиваемым клеточным культурам. Так, например, безуспешными были попытки получить перевиваемые клетки из раковых опухолей желудка и молочных желез человека. С трудом удается адаптировать к жизни invitro клетки плоскоклеточного рака кожи и слизистых. С другой стороны, сравнительно легко выводятся линии из тканей сарком и злокачественных опухолей нервной системы.
Особую категорию клеточных культур составляют полуперевиваемые штаммы, имеющие диплоидный набор хромосом. Они выгодно сочетают в себе черты первичных и перевиваемых клеток.
2. ПИТАТЕЛЬНЫЕ СРЕДЫ.
В любой клеточной культуре различают клеточную и жидкую фазы. Жидкая фаза обеспечивает жизнедеятельность клеток культуры и представляет собой питательные среды различного состава и свойств.
Все среды по своему назначению делятся на ростовые и поддерживающие. В составе ростовых сред должно содержаться больше питательных веществ, чтобы обеспечить активное размножение клеток для формирования монослоя на поверхности стекла или достаточно высокую концентрацию клеточных элементов в суспензии (при получении суспензионных культур). Поддерживающие среды фактически должны обеспечивать лишь переживание клеток в уже сформированном монослое при размножении в клетках вирусных агентов.
Ростовые и поддерживающие среды многокомпонентны. В их состав могут входить как естественные продукты (амниотические жидкости, сыворотки животных), так и субстраты, полученные в результате частичной обработки естественных продуктов (эмбриональные экстракты, гидролизат лактальбумина, гемогидролизат, аминопептид и т. д.), а также синтетические химически чистые вещества (аминокислоты, витамины, соли).
В качестве примера питательной среды, полностью состоящей из естественных компонентов, можно назвать среду Бакли, предложенную для выращивания клеточных культур из почечного эпителия обезьян. В эту среду входит коровья амниотиче-ская жидкость (85%), лошадиная сыворотка (10%) и коровий эмбриональный экстракт (5%).
Все естественные продукты малостандартны, их использование связано с большой опасностью микробной и вирусной контаминации клеточных культур. В связи с этим и происходит постепенное вытеснение их синтетическими стандартными смесями. Наибольшее применение находят синтетическая среда 199 и среда Игла. Широко используются среды, содержащие строго определенные количества солей, аминокислот и витаминов.
Независимо от назначения все питательные среды для тканевых культур конструируются на основе какого-либо сбалансированного солевого раствора с достаточной буферной емкостью. Чаще всего ими являются растворы Хенкса и Эрла. Эти растворы — обязательный компонент любой питательной среды. Неотъемлемым компонентом большинства ростовых сред является сыворотка животных (телячья, бычья, лошадиная), без наличия 5—10% которой размножение клеток и формирование монослоя не происходит.
Включение сыворотки в то же время препятствует созданию ростовых сред точного химического состава, весьма важных для развития фундаментальных исследований по клеточной физиологии, так как вместе с сывороткой (или ее дериватами) вводится целый комплекс неконтролируемых факторов, варьирующих в зависимости от серии сыворотки.
В 50-е годы Ewansи Waymouthбыли предложены бессывороточные среды точного химического состава. Однако эти среды не обеспечивали тех показателей пролиферативной активности клеток, которые обусловливают среды с добавлением сывороток. Всвязи с этимпредставляет интерес работа Birchи Pirt, показавших,что дляобеспечения интенсивногороста клеток в бессывороточных средах определяющим является включение сернокислых солей Fe, Zn, Cu, а также МnС12.
В ростовые питательные среды, а так же в буферный раствор для промывания тканей добавляют антибиотики. Их вводят в среду непосредственно перед употреблением из расчета 1 мл основного раствора антибиотиков на 500 мл среды.
Ниже приведен состав и метод приготовления одной из распространенных питательных сред.
Среда Игла
1. л-аргинина - 17,4
2. л-цистина - 4,8
3. л-гистидина- 3,1
4. л-изолейцина - 26,2
5. л-лейцииа- 13,1
6. л-лизина - 14,6
7. л-метионина - 7,5
8. л-фенилаланина - 8,3
9. л-треонина - 11,9
10. л-триптафана - 2,0
11. л-тирозина - 18,1
12. л-валина - 11,7
13. биотина - 0,24
14. холина - 0,12
15. холин-хлорида - 0,14
16. витамина В12 (птероилглютаминовой кислоты) - 0,44
17. никотинамида- 0,12
18. пантотеновой кислоты - 0,22
19. пантотената кальция - 0,48
20. пиридоксаля (пиридоксин хлоргидрата) - 0,20
21. тиамин-хлоргидрата - 0,34
22. рибофлавина - 0,04
23. хлористого натрия - 5850,0
24. хлористого калия - 373,0
25. фосфата натрия однозамещенного (NaH2PO4 . Н2О) - 138,0
26. кальция хлористого - 111,0
27. двууглекислого натрия (NaHCO3) - 1680,0
28. магния хлористого (MgCl2 . 6Н2О) - 102,0
29. глюкозы - 900,0
30. л-глютамина - 146,2—292,3
31. пенициллина - 50,0
32. стрептомицина - 50,0
33. фенола красного - 5,0
34. водыдо 1000,0
Приготовление.
Раствор 1. В 500 мл воды, нагретой приблизительно до 80°, помешивая, растворяют указанные выше количества аминокислот, после чего добавляют л-глютамин и фенол-красный.
Раствор 2. В 100,0 мл воды растворяют неорганические соли, за исключением двууглекислого натрия (NaHCO3), смешивают с предыдущим раствором неорганических солей и добавляют биотин и витамин B12.
Раствор 3. В 200,0 мл воды растворяют все
витамины, кроме биотина и птеро-илглютаминовой кислоты, и антибиотики —
пенициллин и стрептомицин. Все растворы стерилизуют фильтрованием через
стеклянный фильтр-нутч (пористая стеклянная пластина, величина пор 0,7—1,5 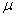
500 мл раствора 1 + 200 мл раствора 2 + 200 мл раствора 3 смешивают и доводят стерильной водой до 1000,0 мл. Можно добавить 1 мг инозита на 1 л.
3. ПОЛУЧЕНИЕ КЛЕТОЧНЫХ КУЛЬТУР.
3.1. ПРИГОТОВЛЕНИЕ ПЕРВИЧНЫХКЛЕТОЧНЫХ КУЛЬТУР.
Первичной - называется культура, полученная из ткани и выращиваемая invitro до начала субкультивирования, то есть до первого посева. Первичная культура лишена многих клеток, присутствующих в исходной ткани, поскольку не все клетки способны прикрепляться к субстрату и выжить invitro. В процессе культивирования клеток происходит относительное обеднение культуры неделящимися или медленно делящимися клетками.
На первом этапе получения первичной культуры проводят стерильное удаление фрагмента ткани, органа животного и его механическую или ферментативную дезагрегацию. Ткань измельчается до кусочков объемом до 1 - 3 мм, кусочки ткани отмываются от эритроцитов раствором Хенкса с антибиотиками. Для дезагрегации ткани используют трипсин (0,25% неочищенный или 0,01 - 0,05% очищенный) или коллагеназу (200 - 2000 ед/мл, неочищенная) и другие протеолитические ферменты. Такой способ получения культуры обеспечивает высокий выход клеток.
Первичные культуры могут быть также получены от кусочков ткани, объемом 1 мм, которые прикрепляются к поверхности субстрата благодаря собственной адгезивности или наличию насечек на чашке, или с помощью сгустка плазмы. В этих случаях будет происходить рост клеток из фрагментов. Клетки, мигрирующие из эксплантатов могут использоваться для пассирования. Фрагменты ткани (эксплантаты) переносятся на новые чашки, мигрирующие клетки могут удаляться пересевом, смесью версена и трипсина, а остающиеся эксплантаты будут образовывать новые выросты.
Известны такие первичные культуры, как культура фибробластов куриного эмбриона, культура клеток почки теленка, лейкоциты.
3.2. ПОЛУЧЕНИЕ МОНОСЛОЙНЫХ ПЕРЕВИВАЕМЫХ КУЛЬТУР КЛЕТОК.
Как уже отмечалось выше, одним из методов получения перевиваемых клеточных линий является отбор клеток с повышенной активностью роста и размножения из популяции первичных культур. Отбор можно осуществить при регулярной смене среды, омывающей монослой первично трипсинизированной клеточной культуры. Для этого отбирают матрасы с хорошо сформированным клеточным монослоем (сами матрасы должны иметь ровное дно и не должны иметь на стенках царапин и матовых пятен). Приготовленную для систематической смены ростовую среду разливают по небольшим сосудам (чтобы исключить бактериальное загрязнение) и хранят при t - 4°.
Смену среды производят регулярно, не реже 1 раза в неделю. В течение первых 3 недель заменяют по 20 - 30% объема ростовой среды, в течение следующих 3 - 4 недель - 50 - 60%, позднее проводят полную смену среды. Свежую среду непосредственно перед работой подогревают до t - 37°.
По мере смены сред клетки меняют свою морфологию. Часть клеток округляется и отпадает от стекла. Большинство клеток стягивается к центру, и монослой приобретает звездчатый вид. Сами клетки при этом несколько удлиняются. Через 7 - 10 смен среды в матрасах, как правило, начинают появляться новые клеточные элементы, причем в разных культурах они имеют разную морфологию.
В культуре клеток почки куриного эмбриона в центре монослоя или между стянутыми его участками появляются округленные клеточные элементы, из которых постепенно формируются скопления в виде небольших колоний. В культуре клеток почки обезьяны появляются одиночные образования, напоминающие зерна. Клетки плотно прилегают друг к другу, образуя мелкие прозрачные колонии. Количество таких клеток нарастает медленно, размеры колоний также почти не увеличиваются. Колонии появляются не только на дне матраса, но также на боковых поверхностях, на границе питательной среды. Поэтому обязательным условием при смене питательной среды является поддержание ее постоянного объема. В культуре клеток почек эмбриона человека на фоне массовой дегенерации стянутого в тяжи монослоя выявляются крупные полигональные клетки с длинными отростками.
Возникшие в процессе отбора атипичные клеточные элементы должны быть отделены от остальных участков монослоя и перенесены в отдельные пробирки или матрасы. Для этого может быть использован метод версенизации или механический откол атипичных клеток с помощью бактериологической петли или шпателя. Последний способ особенно необходим при работе с культурой клеток почек обезьяны, где колонии атипичных клеток плотно прикреплены к стеклу и сами от него не отделяются.
При смене питательной среды в случае появления атипичных клеток рекомендуется осторожно удалить большую часть ростовой среды, а меньшей частью энергично отполоскать монослой и эту среду разлить по пробиркам. В этом случае в среде могут оказаться атипичные клетки, обладающие способностью образовывать колонии, пригодные для дальнейших пересевов.
После переноса культуры атипичных клеток в новую посуду наблюдение за основной культурой целесообразно продолжить, так как процесс выведения новой клеточной линии весьма сложен, и далеко не всегда отобранные атипичные элементы дают начало жизнеспособной линии перевиваемых клеток. Необходимо, чтобы вся работа по получению новых клеточных линий, продолжающаяся в течение многих месяцев, проводилась с одними и теми же питательными средами, сыворотками животных и сериями антибиотиков.
Известны такие первичные культуры, как культура клеток почки золотистого хомячка (BHK), культура клеток почки сибирского горного козерога (ПСГК), культура клеток почки зеленой мартышки (CV).
3.3. РОЛЛЕРНОЕ КУЛЬТИВИРОВАНИЕ КЛЕТОК
До недавнего времени тканевые культуры применялись в вирусологии преимущественно в виде однослойных стационарных культур. Во многих случаях этот метод выращивания клеток является незаменимым. Многолетний опыт показывает, что при использовании однослойных стационарных культур встречается ряд трудностей, связанных с огромными затратами рабочего времени и материалов. С этой точки зрения, более выгодны роллерные культуры, которые экономичны, характеризуются оптимальным отношением полезной площади культивирования к объему питательной среды и открывают благоприятные возможности для накопления клеточной массы.
Термин «роллерные культуры» обозначает метод культивирования, при котором клеточный монослой располагается по всей цилиндрической поверхности горизонтально вращающихся сосудов и периодически омывается питательной средой. В силу определенных причин некоторое количество клеток может в отдельных случаях быть взвешено в культуральной среде. В подобном варианте культуру клеток называют роллерно-суспензионной. "Урожайность" клеточной культуры будет тем больше, чем больше площадь, с которой собирают клетки. Исследователи использовали различные пути увеличения площади поверхности, на которой происходило прикрепление и размножение клеток. Для этого применяли различного характера основы с большой удельной поверхностью: твердую, губчатую, из шерсти, коллагена, на стеклянных спиралях и т. д. Широкого распространения эти методы не получили, так как значительно затрудняли манипуляции с клетками, но следует отметить описанную Л. С. Ратнером и В. А. Крикуном методику многоярусного культивирования. Клетки культивировали в 1,5, 10 и 15-литровых бутылях, полностью загруженных овальными стеклянными трубками, расположенными параллельно продольной оси сосудов. Полезная площадь при этом возрастала по сравнению со стационарными однослойными культурами в сосудах того же объема в 20 раз. Культивирование проходило в 2 этапа. На первом этапе бутыли вращали вокруг горизонтальной оси с целью равномерного распределения клеток. В дальнейшем, когда клетки прикреплялись к стеклу, культивирование продолжалось в стационарном положении. Описанная культуральная система была успешно использована для культивирования вируса ящура.
Более эффективным оказалось вращение сосудов, в которых происходило размножение клеток. Вначале клетки выращивали в пробирках, но с развитием технического оснащения возросли объемы культуральных сосудов, и от выращивания клеток во вращающихся пробирках исследователи перешли к вращающимся бутылям. Роллерные культуры стали применяться как для накопления клеток, так и размножения вирусов.
Для роллерного культивирования используются специальные стеллажно-ярусные аппараты для вращения бутылей или барабаны. Чаще всего для культивирования используют 0,5—3-литровые бутыли. В производственных условиях объем их может достигать 20 литров и более. Аппараты для роллерного культивирования просты, надежны и обслуживание их несложно. Большое значение имеет скорость вращения бутылей. Она не должна быть слишком большой, чтобы не препятствовать прикреплению клеток, но и не слишком низкой, так как длительное нахождение клеток в газовой фазе ухудшает условия их питания. В работах разных авторов скорости вращения бутылей варьировали от 8—12 об/час, до 1 об/мин. Наиболее пригодной для различных клеточных культур оказалась скорость вращения флаконов в пределах 0,5—1 об/мин. Рекомендуется в течение первых 30 мин. после засева клеток обеспечить высокую скорость вращения флаконов (0,5—1,0 об/мин) для равномерного распределения материалов, затем перевести их на низкую скорость вращения (20—30 об/час).
Посевная концентрация в 1 мл среды составляет для клеток СПЭВ, ВНК-21, HeLa, L - 60-80 тыс., для диплоидных клеток 100-200 тыс., для первичных культур - 200-300 тыс. Объем ростовой среды должен составлять 1/10, 1/20 часть объема роллерного флакона. Роллерный метод культивирования позволяет получить большее количество клеток. Так, с флакона емкостью 3 л можно получить урожай клеток HeLa в 8 раз, ВНК-21 - в 9 раз, АО - в 11 раз больший, чем с монослойной стационарной культурой флакона емкостью в 1 л. Кратность экономии сред при использовании 3-литровых флаконов составляет для клеток HeLa 2,8; ВНК-21 - 5,0, АО - 4,0. Способностью к росту и размножению в роллерных условиях обладают субкультуры, перевиваемые культуры и реже первичные, а также диплоидные штаммы клеток. Для лучшего размножения клеток в роллерных аппаратах необходима адаптация их к новым условиям культивирования. Роллерный метод культивирования позволяет получить большие количества клеток. Преимуществом роллерных культур по сравнению с традиционными стационарными культурами является, в частности, более экономичное использование питательных сред и более высокий выход вирусного антигена (на 1—2 lg).
3.4. СУСПЕНЗИОННОЕ КУЛЬТИВИРОВАНИЕ КЛЕТОК
В 1953 году Оуенс с сотрудниками впервые показал способность клеток размножаться в жидкой среде в свободно суспендированном состоянии. С тех пор метод суспензионного культивирования привлекает внимание исследователей в связи с его высокой эффективностью при накоплении больших количеств клеток. Оказалось, что клетки перевиваемых линий, в отличие от остальных типов клеточных культур, могут длительно культивироваться во взвешенном состоянии. Клетки в этих условиях размножаются, не прикрепляясь к стенкам культурального сосуда, находясь в суспензионном состоянии, благодаря постоянному перемешиванию среды. При оптимальном режиме выращивания клетки в суспензиях быстро размножаются, имеют более высокий «урожай», чем в стационарных культурах. Первичные и перевиваемые линии клеток имеют неодинаковую способность роста в суспензии. Так, гетероплоидные и анеуплоидные постоянные линии, в отличие от псевдодиплоидных линий, адаптируются быстрее к росту в суспензии.
Суспензионные культуры готовят из однослойных культур. Клетки отслаивают от стекла с помощью растворов версена и трипсина. Осадок клеток после центрифугирования (1000 об/мин.) ресуспендируют в свежей питательной среде. Приготовленную суспензию помещают в культуральные сосуды (реакторы, ферментеры) и выращивают при постоянном перемешивании. В научных исследованиях концентрация клеток в исходной суспензии колеблется от 0,3 до 10х10 в 1 мл. Оптимальная концентрация клеток в исходной суспензии должна быть 2-5х10 в 1 мл. По мнению Эрла и его сотрудников, концентрация клеток в суспензированной культуре должна быть такой, чтобы фаза логарифмического роста наступала не позднее 16-24 часов после приготовления культуры. Суспензионные клеточные культуры проходят характерные стадии: лаг-фазу, фазу логарифмического роста, стационарную фазу и фазу логарифмического отмирания. В первой стадии, как правило, количество клеток уменьшается, во второй стадии популяция клеток увеличивается (логарифмическая стадия роста), в третьей стадии увеличения количества клеток не наблюдается (стационарная фаза). Если культивирование продолжить дальше, то обнаружится период уменьшения численности клеток-фаза логарифмического отмирания (фаза разрушения). Скорость размножения клеток в логарифмической фазе роста выражают временем генерации. Под временем генерации имеется в виду период, необходимый для удвоения популяции клеток в культуре. Для суспензионного выращивания клеток важным условием является перемешивание жидкости, которое должно быть непрерывным и достаточно интенсивным, чтобы поддерживать клетки во взвешенном состоянии, препятствовать их осаждению и прикреплению к стенкам сосуда и в то же время не вызывать их механического повреждения.
Перемешивание суспензионных культур проводят лопастными магнитными мешалками, а также круговыми качалками. В настоящее время широко применяют вращение флаконов и бутылей вокруг продольной оси (15-40 об/мин). На магнитных мешалках скорость вращения составляет 100—200 об/мин. Скорость перемешивания зависит от объема культуры; малые объемы культуры требуют невысокой скорости, тогда как ее необходимо увеличить при больших объемах. Для предотвращения оседания клеток на внутренней поверхности сосуда производят силиконирование. Силиконовое покрытие в силу своей гидрофобности препятствует прикреплению клеток к стенке сосуда.
Внутренние стенки культурального сосуда смачивают 5% или 10%-ным раствором силикона и после испарения растворителя (бензолового, ацетонового) сосуды выдерживают при 85°С и 200° (соответственно в течение 30 и 60 мин). После охлаждения сосуды наполняют горячей бидистиллированной водой и оставляют на 2 часа, затем их 3 раза ополаскивают бидистиллированной водой и высушивают при 100°С. Сосуды стерилизуют в сушильном шкафу при 170°С в течение 2 часов.
Максимальный рост клеток в суспензии наблюдают при рН 7,0-7,2. Питательные среды, применяемые для выращивания клеток в суспензии, не отличаются от сред, используемых для выращивания клеточных линий в однослойной культуре. Чаще всего при культивировании клеток в суспензиях берут среду Игла с двукратной концентрацией аминокислот и витаминов.
Суспензионные культуры потребляют в 2-7 раз больше глюкозы, чем монослойные. В процессе потребления глюкозы клетки выделяют в среду токсичную для них молочную кислоту. Потребление клетками глюкозы и выработка молочной кислоты идут параллельно и находятся в прямой зависимости от плотности клеточной популяции. Полезным оказалось прибавление к питательной среде инсулина в количестве от 40 до 200 ЕД на 1 л. Прибавление к питательной среде инсулина изменяет соотношение между количеством поглощенной глюкозы и выделенной молочной кислоты. Для клеток линии L указанный коэффициент может быть снижен с 74-81 до 37-38%.
Различные аминокислоты потребляются из питательной среды растущими клетками с неодинаковой скоростью. Отмечено, что регулярное прибавление аргинина (20-40 мг/л) и увеличение количества глутамина до 450 мг/л благоприятствуют росту взвешенных культур.
Прибавление инозитола (0,4 мг/л) позволяет ускорить рост культур амниотических клеток человека. Желательным является добавление к среде каталазы (1 мг/л) и тироксина (12 мг/л).
Большой интерес представляют работы, посвященные получению взвешенных культур клеток в синтетической среде, не содержащей сыворотки крови. Предпринимаются попытки увеличить вязкость питательной среды за счет безбелковых ингредиентов. С этой целью используется метил-целлюлоза и гиалуроновая кислота.
Метилцеллюлоза в концентрации 0,1-0,2% обладает максимальным защитным действием на взвешенные в среде клетки. Протективное действие метилцеллюлозы заключается в том, что молекулы образуют защитный слой вокруг клетки, предотвращающий повреждение клеток при перемешивании среды. Весьма важным показателем состояния суспензионной культуры является парциальное давление кислорода в жидкой фазе. Концентрация кислорода в газовой фазе зависит от плотности клеточной популяции и нередко бывает ниже атмосферной. Недостаток кислорода ведет к появлению грануляции цитоплазмы, клетки теряют правильную округлую форму. При небольшом избытке кислорода клетки имеют хорошо очерченную, правильную, округлую форму, и становятся очень крупными при повреждающем действии избытка кислорода. Оптимальная концентрация кислорода для различных клеточных культур находится в пределах от 9 до 17% или 293 мм рт. столба. При концентрации кислорода выше 20% происходит ингибиция клеточного роста. Так, при концентрации кислорода 24% размножение клеток почек эмбриона кролика (линия ERK) снижалось наполовину, а при 30% сводилось к нулю. Повышение концентрации кислорода токсически воздействует на клеточный метаболизм.
Таким образом, размножение клеток в суспензии зависит от концентрации клеток в исходной суспензии, аэрации и рН среды, состава питательной среды, способа перемешивания, объема суспензии и других факторов.
Однородность суспензии, возможность длительного поддержания клеток в логарифмической фазе роста, перспективы математического моделирования процессов клеточного роста в зависимости от влияния факторов внешней среды, удобство многократного исследования физиологического состояния культуры клеток в суспензии, высокая экономичность метода -вот далеко не полный перечень преимуществ суспензионных культур.
Суспензионные культуры широко используется в вирусологических исследованиях и для накопления больших количеств вируссодержащего материала, при изготовлении вакцин и диагностических препаратов.
3.5. КУЛЬТИВИРОВАНИЕ КЛЕТОК НА МИКРОНОСИТЕЛЯХ.
В 1967 г. VanWerel предложил метод культивирования, сочетающий элементы монослойного и суспензионного выращивания клеток, который он назвал методом «микроносителей». Суть его заключается в том, что клетки прикрепляются и размножаются на поверхности полимерных шариков-частиц «микроносителей» (МН), которые содержатся в суспензии с помощью перемешивающего устройства, например мешалки. На одной частице МН диаметром 160—230 мм может поместиться 350-630 (или в среднем 460) клеток. В одном мл среды можно суспензировать несколько тысяч частиц микроносителя, при этом общая площадь их составит от нескольких до 50 см2/мл.
Инокулированные в культиватор клетки прикрепляются к поверхности частиц МН и размножаясь, образуют сплошной монослой на каждой отдельной частице.
Основными преимуществами этого метода являются:
1) создание равномерных условий по всему объему сосуда, что делает возможным эффективно контролировать необходимые параметры (рН, р02 и др.); 2) получение высокой плотности клеточной популяции до 5-6 млн. клеток в 1 мл; 3) культивирование одновременно несколько сот миллиардов клеток; 4) введение постоянного контроля за динамикой роста клеток; 5) снижение роста контаминации в связи с сокращением операций, связанных с разгерметизацией культураль-ного сосуда; 6) значительная экономии питательных сред; 7) возможность сохранять выросшие клетки непосредственно на частицах при низких температурах; 8) возможность искусственно создавать различные концентрации МН с выросшими на них клетками; 9) возможность пассирования культуры без применения трипсина путем добавления свежих порций микроносителя.
Микроносители должны иметь:
- небольшой положительный заряд в пределах 1,5-1,8 МЭКВ/г. В связи с тем, что большинство клеток животных имеют слабо отрицательный заряд, они легче будут прикрепляться к такому МН:
- плотность 1,05—1,15 г/см; указанная плотность является оптимальной для поддержания МН во взвешенном состоянии;
- диаметр частиц от 100 до 250 мкм, что обеспечивает площади для роста нескольких сотен клеток;
- гладкую поверхность;
- прозрачность;
- отсутствие токсичности компонентов для клеток;
- незначительное впитывание компонентов среды;
- универсальность, обеспечивающую возможность использования их для первичных, диплоидных и гетероплоидных клеток. Немаловажное значение имеют свойства МН, которые позволяют использовать их многократно.
Проведено исследование многих гранулированных препаратов различной химической природы, в том числе из поперечно-сшитого (ПС) декстрана, ПС-агарозы, ПС-поливинилпиролидона, полиакрилнитрита, пористого селикагеля, полистирола, капрона, нейлона, алюмосиликата с целью использования их как микроносителей.
Пригодными являются только некоторые из них, главным образом имеющие в своей основе ПС-декстран.
Несколько зарубежных фирм разработали коммерческие препараты микроносителей, готовые к употреблению: Цитодекс-1, 2, 3 (Франция, Швеция), Супербит (США, Англия), Биосилон (Дания). Стоимость перечисленных препаратов довольно высока, поэтому необходимо проводить исследования по разработке и производству отечественных МН.
Культивирование клеток на микроносителях проводят в обычных ферментерах для суспензионного культивирования. В ферментере не должно быть каких-либо выступов, карманов, чтобы предотвратить накопление микроносителей в застойных зонах. Поэтому разумно использовать ферментеры с круглым дном и гладкими стенками. Внутренняя поверхность ферментера должна быть силиконизирована для предотвращения прилипания микроносителей к стеклу или нержавеющей стали ферментера.
Для контроля за окружающими культуру условиями такими, как рН и О2, может быть использовано стандартное оборудование. Скорость перемешивания суспензии с носителем должна быть 40-60 об/мин. Для выращивания на МН применяются различные типы клеток.
Концентрация цитодекса может варьировать от 0,5 до 5 мг/мл. Однако, при производстве профилактических вакцин, применяют обычно конечную концентрацию цитодекса, не превышающую 1 мг/мл. Повышение концентрации до 3 мг/мл и выше создает дополнительные трудности, связанные с необходимостью перфузии питательной среды и частичной ее замены, что осложняет технологический процесс.
Посевная концентрация клеток, а также условия культивирования на МН в первые часы в значительной степени определяют оптимальные параметры для пролиферации и максимального накопления клеток. Показано, что посев 10 клеток/мл перевиваемой линии почек обезьян (Vero) и диплоидных клеток фибробластов эмбриона человека (MRC-5) в уменьшенном до 1/3 объема питательной среды и при периодическом включении мешалки (30 об/мин) на одну минуту через каждый час в течение 4 часов, с последующим добавлением питательной среды до конечного объема, ведет к увеличению пролиферации клеток и их количества по сравнению с контролем (полный объем питательной среды в момент посадки клеток и непрерывная работа мешалки с начала культивирования).
Важное значение для культивирования клеток на МН имеет питательная среда. Правильный подбор питательной среды также будет способствовать оптимизации процесса пролиферации клеток и их качество. Необходимо проводить подбор питательных сред для культивирования клеток на различных типах МН. Показано, что перевиваемая линия клеток почек обезьян (Vero) на цитодексе дает наибольший выход при использовании среды Игла ДМЕ (посадочная концентрация клеток 105 мл) но сравнению со средой ВМЕ и 199. Если же количество клеток при посадке снизить до 104, то лучшие результаты дает среда 199. Все испытуемые среды содержали 10% фетальной сыворотки.
4. ВЫРАЩИВАНИЕ ВИРУСОВ В КУЛЬТУРАХ КЛЕТОК.
В настоящее время для выделения и размножения вирусов животных используются первичные культуры, штаммы клеток и установившиеся клеточные линии. В общих чертах процедура оказывается одинаковой для всех вирусов.
Среду удаляют с клеточного монослоя и монослой промывают сбалансированными буферными солевыми растворами (СБСР) или фосфатно-солевым буфером (ФСБ) для удаления ингибиторов (антител), которые могут присутствовать в среде. Вирусные частицы суспендируются в небольшом количестве СБСР или ФСБ и адсорбируются клетками в течение 30 - 60 мин. После этого солевые растворы заменяются свежей средой.
Заражение вирусами культивируемых клеток вызывает характерные морфологические изменения клеток. Конечные дегенеративные клеточные процессы (цитопатогенный эффект, ЦПЭ) обнаруживаются только через несколько недель роста в присутствии вирусов, но в ряде случаев ЦПЭ обнаруживаются уже через 12 ч. Детали морфологических изменений оказываются различными в случае разных вирусов.
Если вместо продуктивной инфекции вирус вызывает клеточную трансформацию, то это также сопровождается характерными изменениями морфологии и особенностей роста клеток.
4.1. ПРЕДОСТОРОЖНОСТИ ПРИ РАБОТЕ С ЗАРАЖЕННЫМИ ВИРУСАМИ КЛЕТКАМИ.
Вирусы оказывают цитопатогенное действие и служат этиологическими агентами при многих заболеваниях человека и животных. Кроме того, многие вирусы (например, онкорнавирусы, вирус герпеса тип II, аденовирусы, вирус полиомы и SV40) являются, по-видимому, агентами, вызывающими развитие опухолей у животных. Из-за способности вирусов проходить через бактериальные фильтры бывает трудно исключить вирусы из культур незараженных клеток при наличии вирусных суспензий, когда возможна передача вируса через воздух культуральной комнаты.
Указанные ниже меры предосторожности носят самый общий характер и применимы только в тех случаях, когда вирусы не представляют какой-либо особой опасности. При использовании особо опасных вирусов, представляющих опасность для здоровья сотрудников лаборатории или людей и животных окружающего мира, следует применять дополнительные меры предосторожности. К вирусам, представляющим особую опасность, относятся вирусы ньюкаслской болезни, вирусы ящура, вирусы везикулярного стоматита, вирусы оспы, вирусы бешенства, вирусы герпеса типа В и так далее. Кроме того, нельзя быть уверенным, что даже такие вирусы, как SV40, не представляют опасности для человека.
1) Для заражения клеток и роста зараженных вирусами клеток должна использоваться специальная комната или группа комнат.
2) Никакие живые вирусы не должны выноситься из этих помещений, не будучи заключены в плотно закрывающиеся контейнеры. Следует принимать меры предосторожности, чтобы наружные поверхности этих контейнеров не были загрязнены вирусными частицами.
3) При работе с вирусами должны быть использованы специальные защитные одежды (лабораторные халаты). После работы эта одежда должна помещаться в специальный бак для автоклавирования.
4) Все среды и стеклянная посуда, которые были в контакте с вирусами, должны быть обработаны хлоросом; только после этого их можно выносить из помещения, предназначенного для работы с вирусами.
5) Вся пластиковая посуда должна быть помещена в специальный бак для автоклавирования.
6) Оборудование для хранения и обработки вирусного материала должно размещаться внутри помещения для работы с вирусами.
7) Лаборатория должна быть оснащена двухцикловыми автоклавами, чтобы сотрудники лаборатории были защищены от вредоносных материалов.
4.2. ОБНАРУЖЕНИЕ ВИРУСОВ.
Обнаружить присутствие вирусов и определить их количество можно с помощью целого ряда различных тестов, например:
1) Активность вируса измеряется путем определения количества содержащего вирус материала, необходимого для того, чтобы вызвать специфическую реакцию в организме хозяина. Индуцируемая вирусом реакция может происходить по типу «все или ничего» (т. е. наличие или отсутствие инфекции), а может быть выражена количественно, например продолжительностью времени, необходимого для проявления инфекции, или числом поражений в слое чувствительных клеток. Количественное определение вирусной активности называется титрованием.
Наименьшее количество вируса, способное вызвать соответствующую реакцию, называется инфекционной единицей, и титр исходной вирусной суспензии выражается числом инфекционных единиц, приходящихся на единицу объема. Например, если минимальное количество суспензии вируса гриппа, вызывающее у мыши пневмонию при интраназальном введении суспензии ткани инфицированного легкого в разведении 1:106 равно 0,1 мл, то это значит, что титр вируса гриппа в исходной суспензии ткани легкого равен 107 инфекционным единицам на 1мл—1/(10~1-10-6) =107.
Как правило, обязательным компонентом метода титрования вируса является тест, позволяющий оценить результат инокуляции как положительный или отрицательный. Например, при титровании вирусов животных таким тестом может быть наличие или отсутствие видимых поражений в культуре клеток, воспалительная реакция на месте инокуляции вируса в кожу или роговицу, параличи, возникающие у животного в результате введения вируса в мозг, и т. д. Иногда размножение вируса можно выявить и в отсутствие видимой реакции со стороны организма-хозяина: на пример, заражение куриных эмбрионов миксовирусами можно обнаружить по появлению гемагглютининов в аллантоисной жидкости.
Наилучшие результаты дают методы титрования, в основе которых лежит подсчет числа дискретных поражений на определенной площади слоя клеток, зараженных известным количеством содержащего вирус материала. В случаях когда число чувствительных к вирусу и доступных для него клеток можно практически считать бесконечно большим, как, например, при заражении фагом однослойной культуры бактерий на питательном агаре или при заражении вирусом сплошной однослойной культуры животных клеток, поражения, вызываемые вирусом, или бляшки, образуемые фагом, в данном смысле подобны колониям бактерий на плотной питательной среде. Как число этих колоний пропорционально числу бактериальных клеток, содержащихся в титруемом препарате, так и число бляшек прямо пропорционально количеству вируса, добавленному к однослойной культуре образование зараженными клетками вирусных частиц, которые могут быть идентифицирован, например, по природе нуклеиновых кислот и симметрии частиц.
2) Образование зараженными клетками нуклеиновых кислот, которые могут обладать характерной плотностью при центрифугировании в градиенте плотности или характерным размером при электрофорезе в агарозном геле или центрифугировании в градиенте концентрации сахарозы.
3) Образование зараженными клетками или трансформированными клетками характерных антигенов, которые могут быть визуализованы с помощью окрашивания флуоресцирующими специфическими антителами либо вызывать изменения в клеточной мембране, приводящие к адсорбции гема. В прямом методе антитела, полученные к вирусным антигенам, сочетаются с флуорохромом и используются для окраски зараженных клеток. Положительная реакция (желто-зеленый цвет в люминесцентном микроскопе) указывает на присутствие в клетках вирусных антигенов. Этот метод, следовательно, может быть использован для выявления клеточной трансформации, когда антитела вырабатываются, например, против ранних антигенов вируса SV40, или для выявления образования зрелых вирусов, когда получаются антитела против капсидных белков. В непрямом методе антитела не сочетаются с флуорохромом. Вместо этого после взаимодействия антител с антигенами в фиксированных препаратах клеток к ним добавляются антигамма-глобулиновые антитела, конъюгированные с флуорохромом. Этот метод более чувствителен и позволяет избежать конъюгации каждого индивидуального антитела с флуоресцентным красителем. Так, одни и те же флуоресцентные антитела к антителам кролика (полученные у овец против кроличьих гамма-глобулинов) могут быть использованы для окраски любых антител, образующихся у кроликов и взаимодействующих с вирусными антигенами в продуктивно зараженных или трансформированных клетках. Еще более непрямой, но значительно более чувствительный метод, не требующий использования флуоресцентного микроскопа - пероксидазный - антипероксидазный метод. Согласно этому методу, кроличьи антитела взаимодействуют с антигенами фиксированных клеток и затем покрываются антителами козы к иммуноглобулинам кролика, конъюгированными с комплексами пероксидазы хрена и кроличьей антипероксидазы. После этого препараты обрабатывают диамино-бензидином и перекисью водорода; коричневая окраска указывает на присутствие антигенов.
4) Наличие антигенов на вирусных частицах может приводить к реакциям гемагглютинации, позволяющим проводить количественную оценку. У многих вирусов имеются антигены, которые могут адсорбироваться на эритроцитах и вызывать их слипание. При взаимодействии большого количества вирусных частиц и эритроцитов образуется сеть клеток, и суспензия агглютинирует, т.е. эритроциты выпадают в осадок. Существует некоторая специфичность, заключающаяся в том, что определенные вирусы вызывают агглютинацию эритроцитов определенных животных, но если обнаружена активная комбинация, то гемагглютинация становится быстрым тестом, позволяющим определить титр вируса. Кровь отбирают в гепаринизированный шприц и эритроциты промывают трехразовым переосаждением в 0,85%-ном NaCl при 200 gв течение 10 мин. Окончательный осадок эритроцитов суспендируют в 200-кратном объеме 0,85%-ного раствора NaCl. Удобнее всего проводить тест на гемагглютинацию в микротитровальных пластинках. В серию лунок микротитровальной пластинки вносят по 25 мкл 0,85%-ного NaClили ФСБ; в первую лунку вносят 25 мкл суспензии вируса, и смесь перемешивают (разведение 1:2). 25 мкл переносят затем из первой лунки во вторую (разведение 1:4) и так далее, до конечного разведения 1:2048. После этого в каждую лунку добавляют по 25 мкл суспензии эритроцитов, смесь перемешивают и оставляют при 4°С, комнатной температуре или 37 °С до наступления гемагглютинации (1-2 ч). В лунках, где произошла агглютинация, осадок эритроцитов имеет неправильную форму, а в лунках, где гемагглютинации не было, клеточный осадок образует компактную точку на дне лунки. Разведение в последней лунке, в которой происходила гемагглютинация, рассматривается как титр, и этому разведению приписывается значение 1 гемагглютинирующей единицы (ГАЕ). Предшествующее разведение содержит соответственно 2 ГАЕ и так далее.
5) Образование зараженными клетками характерных ферментов, или ферментов, легко отличаемых по свойствам от соответствующих ферментов клеток-хозяев.
4.3. КУЛЬТИВИРОВАНИЕ ПИКОРНАВИРУСОВ НА ПРИМЕРЕ ПОЛУЧЕНИЯ ПОЛИОВИРУСА ТИПА 3 В КУЛЬТУРЕ КЛЕТОК HeLa.
В качестве конкретной методики культивирования вирусов можно привести способ выращивания полиовируса в перевиваемых клетках.
Все процедуры проводятся в стерильных условиях.
1. Определенные сублинии клеток HeLa(например, HeLaS3) могут расти в средах с низкой концентрацией ионов кальция (например, CaS2MEM). Для эффективного заражения существенно, чтобы клетки хорошо росли в виде однородной суспензии. Клетки осаждают низкоскоростным центрифугированием (15 мин при 900 g) и ресуспендируют в среде CaS2MEM до концентрации ~ 2.107 кл/мл (~1/20 исходного объема).
2. К клеточной суспензии добавляют вирус в концентрации 10—50 БОЕ на клетку; инкубируют ее на магнитной мешалке в течение 1 ч при 35 °С.
3. Суспензию разводят той же средой до концентрации 2.106 кл/мл и добавляют 100 мкКи [3Н]-уридина.
4. Клеточную суспензию инкубируют в течение 8 ч, после чего осаждают клетки низкоскоростным центрифугированием (15 мин при 900 g) и промывают один раз фосфатным буферным раствором (PBS), не содержащим ионов магния и кальция.
5. Клетки ресуспендируют в PBS (5-107кл/мл) и проводят три цикла замораживания — оттаивания.
6. Остатки клеток удаляют центрифугированием.
7. Супернатант сохраняют для дальнейшей очистки.
СПИСОК ЛИТЕРАТУРЫ
1. Адамс Р. Методы культуры клеток для биохимиков. - М.: Мир, 1983, 263 с.
2. Вирусология. Методы. / Под ред. Б. Мейхи. - М.: Мир, 1988, 344 с.
3. Голубев Д. Б., Соминина А. А., Медведева М. Н. Руководство по применению клеточных культур в вирусологии. - Л.: Медицина, 1976, 224 с.
4. Дьяконов Л. П., Глухов В. Ф., Поздняков А. А., Денисенко Г. Ф., Калмыкова Т. П. Культивирование клеток и тканей животных. - Ставрополь: Ставроп. Правда, 1988, 2 часть, 91 с.
5. Животная клетка в культурепод. ред. Л. П. Дьяконова, В. И. Ситькова. – М.: 2000
6. Культура животных клеток. Методы. / Под ред. Р. Фрешни. - М.: Мир, 1989, 333 с.
7. Руководство по ветеринарной вирусологии. / Под ред. В. Н. Сюрина. - М.: Колос, 1965, 687 с.
8. Сергеев В. А. Репродукция и выращивание вирусов животных. - М.: Колос, 1976, 303 с.
9. Феннер Ф., Мак-Ослен Б., Мимс С., Сэмбрук Дж., Уайт Д. Биология вирусов животных. - М.: Мир, 1977, т. 1, 447 с.